Síndrome de Hunter: Una Enfermedad de Almacenamiento Lisosomal
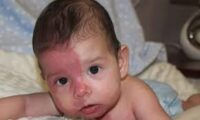
El Síndrome de Hunter, también conocido como Mucopolisacaridosis II (MPS II), es un trastorno genético raro que pertenece al grupo de las mucopolisacaridosis. Esta enfermedad afecta el metabolismo de los glicosaminoglicanos (GAGs), lo que provoca su acumulación en diferentes órganos y tejidos del cuerpo. El síndrome es más común en niños varones y tiene una herencia ligada al cromosoma X.
Causas
El Síndrome de Hunter es causado por una mutación en el gen IDS, que produce la enzima iduronato-2-sulfatasa. Esta enzima es responsable de descomponer ciertos tipos de GAGs, como el dermatán sulfato y el heparán sulfato. La deficiencia de esta enzima provoca la acumulación de estos compuestos en las células y tejidos, lo que interfiere con el funcionamiento normal de varios órganos.
Síntomas
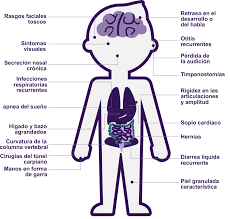
Los síntomas pueden variar en gravedad, pero suelen comenzar entre los 2 y 4 años de edad. Los principales síntomas incluyen:
• Rasgos faciales toscos (nariz ancha, labios gruesos, frente prominente).
• Hernias (umbral o inguinales).
• Retraso en el desarrollo y dificultades cognitivas.
• Hiperactividad y agresividad.
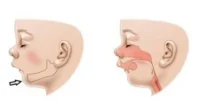
• Problemas respiratorios, como apnea del sueño y dificultad para respirar.
• Discapacidad intelectual (en algunos casos severa).
• Rigidez articular y problemas ortopédicos (como escoliosis).
• Problemas cardíacos, como enfermedades valvulares o dilatación del corazón.
• Pérdida de audición y visión.
• Aumento del tamaño del hígado y el bazo (hepatoesplenomegalia).
• Síntomas en la piel, como engrosamiento de la dermis.
Diagnóstico
El diagnóstico del Síndrome de Hunter incluye:
• Evaluación clínica de los síntomas observados.
• Análisis de orina para detectar los GAGs acumulados.
• Pruebas enzimáticas para medir la actividad de la iduronato-2-sulfatasa.
• Estudios genéticos para identificar la mutación en el gen IDS.
Tratamiento
Aunque no existe una cura para el Síndrome de Hunter, existen varias opciones terapéuticas para controlar los síntomas y mejorar la calidad de vida de los pacientes:
• Terapia Enzimática Sustitutiva (TES): La administración de idursulfasa, que reemplaza la enzima deficiente y ayuda a reducir la acumulación de GAGs.
• Trasplante de células madre hematopoyéticas (TCMH): Este tratamiento puede ser útil si se realiza en etapas tempranas de la enfermedad.
• Cirugías: Para tratar algunas complicaciones ortopédicas, cardíacas o respiratorias.
• Fisioterapia y terapia ocupacional: Para mejorar la movilidad y la calidad de vida.
• Tratamiento para la pérdida auditiva y visual: Monitoreo y corrección de problemas de oído y vista.
Pronóstico
El pronóstico varía según la gravedad de los síntomas y el momento en que se inicia el tratamiento. Si no se trata, el Síndrome de Hunter puede reducir significativamente la esperanza de vida. Sin embargo, con un manejo adecuado, algunos pacientes pueden tener una calidad de vida mejorada y una mayor esperanza de vida.