Síndrome de Apert – Malformación Craneofacial Congénita
¿Qué es?
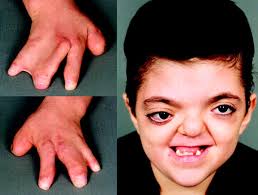
El síndrome de Apert es un trastorno genético raro que causa fusión prematura de los huesos del cráneo (craneosinostosis), así como malformaciones en cara, manos y pies. Esta fusión impide el crecimiento normal del cráneo y puede afectar el desarrollo cerebral y facial.
Causa
- Mutación en el gen FGFR2 (receptor del factor de crecimiento de fibroblastos).
- La mayoría de los casos son nuevas mutaciones, no heredadas.
- Cuando se hereda, tiene un patrón autosómico dominante.
Características principales
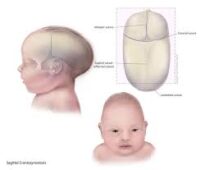
- Craneosinostosis:
- Fusión temprana de suturas craneales.
- Cráneo alto y abombado (braquicefalia).
- Frente prominente y cara plana.
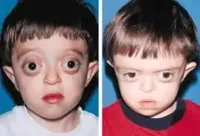
- Malformaciones faciales:
- Hipoplasia mediofacial (puente nasal deprimido).
- Ojos prominentes y separados (hipertelorismo).
- Problemas dentales y paladar hendido (en algunos casos).
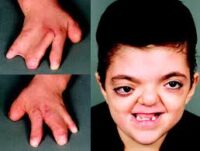
- Sindactilia (fusión de dedos):
- Dedos de manos y pies fusionados (como “manos en mitón”).
- Puede afectar la funcionalidad de las extremidades.
Otros síntomas posibles
- Retraso en el desarrollo (variable).
- Problemas respiratorios.
- Infecciones de oído frecuentes.
- Problemas visuales o auditivos.
- Hidrocefalia en algunos casos.
Diagnóstico
- Clínico, basado en las características físicas.
- Confirmación genética con prueba del gen FGFR2.
- Estudios de imagen: radiografías, tomografía o resonancia.
Tratamiento
- Cirugía craneal temprana para evitar presión intracraneal y permitir crecimiento cerebral.
- Cirugías reconstructivas faciales.
- Corrección quirúrgica de manos y pies.
- Apoyo multidisciplinario: neurocirujano, genetista, ortopedista, logopeda, psicólogo, etc.
Pronóstico
- No tiene cura, pero con tratamiento adecuado, muchos niños logran una buena calidad de vida.
- El pronóstico depende de la severidad de las malformaciones y de la intervención temprana.