Síndrome de Pfeiffer: una craneosinostosis de origen genético
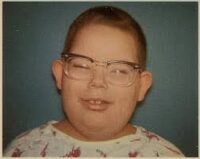
El Síndrome de Pfeiffer es una enfermedad congénita poco común que forma parte de los síndromes de craneosinostosis, caracterizados por el cierre prematuro de las suturas craneales. Este cierre impide el desarrollo normal del cráneo, y se asocia con anomalías faciales y malformaciones en manos y pies.
Causa genética
El síndrome es causado por mutaciones en los genes FGFR1 y FGFR2 (Receptores del Factor de Crecimiento de Fibroblastos tipo 1 y 2). Se hereda con un patrón autosómico dominante, aunque también pueden presentarse casos esporádicos por mutaciones nuevas sin antecedentes familiares.
Tipos clínicos
El Síndrome de Pfeiffer se clasifica en tres tipos, dependiendo de la gravedad de las manifestaciones:
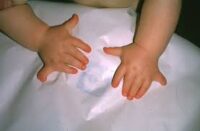
- Tipo 1 (clásico): es la forma más leve. Los pacientes presentan craneosinostosis, frente prominente, hipertelorismo (ojos separados), y dedos anchos o en forma de espátula, tanto en manos como pies. La inteligencia suele ser normal.
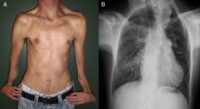
- Tipo 2: se asocia con craneosinostosis severa con forma de “trébol” en el cráneo (cráneo cloverleaf), proptosis ocular (ojos muy prominentes), y malformaciones severas en extremidades. Puede haber retraso mental y problemas respiratorios.
- Tipo 3: similar al tipo 2, pero sin cráneo en trébol. También se asocia con afectación neurológica severa, malformaciones viscerales y alta mortalidad.
Características clínicas
- Cierre prematuro de suturas del cráneo (principalmente coronal).
- Frente amplia y forma anómala del cráneo.
- Ojos prominentes y separados (exoftalmos e hipertelorismo).
- Hipoplasia mediofacial (cara plana).
- Dedos de manos y pies cortos y anchos (braquidactilia).
- Malformaciones auditivas y problemas respiratorios.
- En los tipos severos, puede haber hidrocefalia, convulsiones y retraso del desarrollo.
Diagnóstico
Se realiza mediante evaluación clínica, estudios por imagen (como tomografía craneal) y pruebas genéticas para identificar mutaciones en FGFR1 o FGFR2. Puede diagnosticarse prenatalmente mediante ecografías y análisis genéticos si hay antecedentes familiares.
Tratamiento
El manejo es multidisciplinario, e incluye:
- Cirugías craneofaciales para corregir deformidades y prevenir presión intracraneal.
- Intervenciones quirúrgicas en manos y pies, si hay limitación funcional.
- Monitoreo neurológico, oftalmológico y respiratorio.
- Terapia del lenguaje, física y ocupacional, en los casos que lo requieran.
Pronóstico
- El tipo 1 tiene un buen pronóstico, especialmente con intervenciones oportunas.
- Los tipos 2 y 3 tienen un pronóstico reservado, con alta mortalidad en los primeros años de vida, especialmente si no se trata adecuadamente.