Síndrome de Carpenter: malformaciones craneales, digitales y más
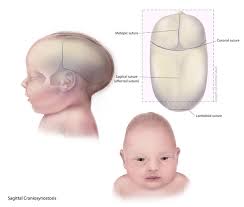
El Síndrome de Carpenter, también conocido como acrocefalopolisindactilia tipo II, es una enfermedad genética rara que afecta el desarrollo del cráneo, la cara, las extremidades y, en algunos casos, el funcionamiento intelectual. Pertenece al grupo de las craneosinostosis sindrómicas, donde las suturas del cráneo se cierran antes de tiempo, afectando la forma de la cabeza y el crecimiento cerebral.
Causa genética
Este síndrome es causado principalmente por mutaciones en el gen RAB23, que se transmite con un patrón de herencia autosómico recesivo. Para que un niño lo desarrolle, debe heredar una copia defectuosa del gen de ambos padres. En casos más raros, también puede haber implicación de otros genes como MEGF8.
Manifestaciones clínicas
El Síndrome de Carpenter presenta una combinación de malformaciones físicas y, en algunos casos, retraso en el desarrollo. Los signos más comunes incluyen:
• Craneosinostosis, especialmente de la sutura coronal, que produce una forma puntiaguda del cráneo (acrocefalia).
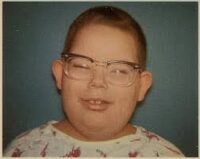
• Facies característica, con cara plana, hipertelorismo (ojos separados), y nariz pequeña.
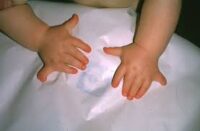
• Sindactilia (dedos unidos), común en manos y pies.
• Polidactilia (dedos adicionales), especialmente en los pies.
• Obesidad infantil precoz.
• Retraso en el desarrollo psicomotor e intelectual, en muchos casos.
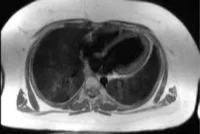
• Anomalías cardíacas congénitas y otras malformaciones internas, como renales o genitales.
• Hipogenitalismo en varones.
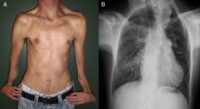
• Baja estatura y talla por debajo del promedio.
Diagnóstico
El diagnóstico suele hacerse mediante la evaluación clínica detallada, apoyado por radiografías que muestran la craneosinostosis y las malformaciones óseas. También puede confirmarse mediante pruebas genéticas moleculares para identificar la mutación en el gen RAB23 o MEGF8.
Tratamiento
El manejo del síndrome es multidisciplinario, enfocado en mejorar la calidad de vida del paciente:
• Cirugías craneofaciales para corregir la forma del cráneo y prevenir aumento de presión intracraneal.
• Corrección quirúrgica de polidactilia y sindactilia, para mejorar la función de las manos y pies.
• Monitoreo del desarrollo intelectual y apoyo con educación especial si es necesario.
• Atención cardiológica, nefrológica y endocrina, según las malformaciones presentes.
• Asesoramiento genético a las familias, ya que el riesgo de recurrencia es alto si ambos padres son portadores.
Pronóstico
El pronóstico varía según la gravedad de las malformaciones y el acceso a tratamiento. Aunque pueden existir complicaciones importantes, muchos niños con intervención temprana y cuidados adecuados pueden alcanzar una buena calidad de vida. Sin embargo, el retraso intelectual y los problemas médicos asociados pueden requerir apoyo permanente.