Síndrome de Holt-Oram: Trastorno Genético Cardíaco y de Extremidades Superiores
El síndrome de Holt-Oram es una enfermedad genética autosómica dominante que afecta principalmente el desarrollo de los miembros superiores (especialmente los brazos y manos) y el corazón, provocando malformaciones esqueléticas y anomalías cardíacas congénitas.
Características principales:
1.
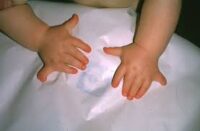
Alteraciones esqueléticas
:
- Afectan principalmente los dedos, muñecas, antebrazos y hombros
- Pulgares ausentes, subdesarrollados o en forma de dedo (dedo trifalángico)
- Fusión o anomalías en los huesos del carpo
- Brazos cortos o asimetría en la longitud de las extremidades
- Suele afectar un solo lado, pero puede ser bilateral
2.
Anomalías cardíacas
:
- Comunicación interauricular (CIA) o interventricular (CIV)
- Arritmias cardíacas, incluso en ausencia de malformaciones estructurales
- En casos más severos: bloqueos cardíacos o necesidad de marcapasos
Causas:
- Mutación en el gen TBX5, ubicado en el cromosoma 12
- Se hereda de forma autosómica dominante, es decir, una sola copia del gen alterado puede causar la enfermedad
- También pueden darse casos nuevos (de novo) sin antecedentes familiares
Diagnóstico:
- Evaluación clínica de las malformaciones esqueléticas
- Electrocardiograma (ECG) y ecocardiograma para detectar anomalías cardíacas
- Estudios genéticos para confirmar la mutación en el gen TBX5
- Diagnóstico prenatal posible mediante ecografías y pruebas genéticas
Tratamiento:
- Cirugías ortopédicas para mejorar la funcionalidad de los miembros superiores
- Rehabilitación y terapia ocupacional
- Monitoreo cardiológico frecuente
- En caso de cardiopatías severas:
- Cirugía correctiva cardíaca
- Marcapasos si hay arritmias importantes
Pronóstico:
- Varía según la severidad de las malformaciones
- Con tratamiento adecuado, los pacientes pueden llevar una vida funcional y activa
- Es fundamental un seguimiento cardiológico de por vida